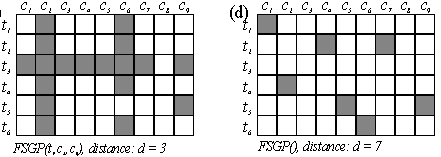|
cGDM : clinical Genome Data Model
In the light of recent developments in genomic technology and rapid accumulation of genomic information, a major transition toward precision medicine is anticipated. However, the clinical applications of genomic information remain limited. This lag can be attributed to several complex factors, including the knowledge gap between medical experts and bioinformaticians, the distance between bioinformatics workflow and clinical practice, and the unique characteristics of genomic data, which can make interpretation difficult. We aim to present a novel genomic data model, which allows for more interactive support in clinical decision-making. Informational modelling was used as base knowledge to design the communication scheme between sophisticated bioinformatics prediction and the representative data of a clinical decision. This study was conducted by a multidisciplinary working group who carried out clinico-genomic workflow analysis and attribute extraction, through Failure Mode and Effects Analysis. Based on those results, a clinical genome data model (CGDM) was then developed with eight entities and 46 attributes. The CGDM encompasses reliability-related factors that enable clinicians to access the reliability problem of each genomic test result as clinical evidence. The proposed CGDM could serve as a data-layer infrastructure supporting the intellectual interplay between medical experts and informed decision-making.
RarePedia: A knowledge base for biomedically coherent query result

For making use of the results provided by public biomedical research, we developed a biomedical knowledgebase, RarePedia, as well as a web portal for query service. RarePedia presents a coherent query result in accordance with central dogma scheme. For a variant or gene information query, RarePedia presents variant, gene, transcript, protein, pathway, and phenotype section web pages. Especially, variant and phenotype sections will provide pivotal information for genetic counselors or laboratory physicians in the molecular pathology field by presenting important information but time-consuming when it is searched in different databases. RarePedia also presents the information described above through REST API to support computer system oriented rather than human oriented bioinformatical information retireval.
CaReAl: Capturing Read Alignments in a BAM file rapidly and conveniently
![]() For the systematic exploration of the overall read-alignment status!
For the systematic exploration of the overall read-alignment status!
CaReAl [kæriːəl] is a high-performance alignment capturing tool for visualizing the read-alignment status of nucleotide sequences and associated genome features. This rapid user-programmable capturing tool is useful for obtaining read-level data for evaluating variant calls and detecting technical biases.
MELLO: Medical Lifelog Ontology
 Every daily activity may be logged by MELLO!
Every daily activity may be logged by MELLO!
MELLO web services with MELLO Browser and MELLO Mapper helps Neo for logging his life activities.
RCARE: RNA-Seq Comparison and Annotation of RNA Editing
 The RNA world will be monitored and editted in every detail
The RNA world will be monitored and editted in every detail
Post-transcriptional sequence modification of transcripts through RNA editing is an important mechanism for regulating protein function and correlated with human disease phenotypes. Identifying RNA editing sites is the basic step for studying RNA editing. Determining condition-specific RNA editing sites and elucidating their functional roles help understanding various biological phenomena mediated by RNA editing. We propose RCARE (RNA-Seq Comparison and Annotation for RNA Editing) that searches, annotates and visualizes RNA editing sites using previously known thousands of editing sites. RCARE performs comparative analysis between multiple samples.
hiHMM: Bayesian non-parametric joint inference of chromatin state maps
 Kyung-Ah Sohn, Joshua Ho, Djordje Djordjevic, Peter J. Park and Ju Han Kim
Kyung-Ah Sohn, Joshua Ho, Djordje Djordjevic, Peter J. Park and Ju Han Kim
hiHMM (hierarchically-linked infinite hidden Markov model) is a new Bayesian non-parametric method to jointly infer chromatin state maps in multiple genomes (different cell types, developmental stages, even multiple species) using genome-wide histone modification data.
Bien's measure of semantic similarity for Gene Ontology
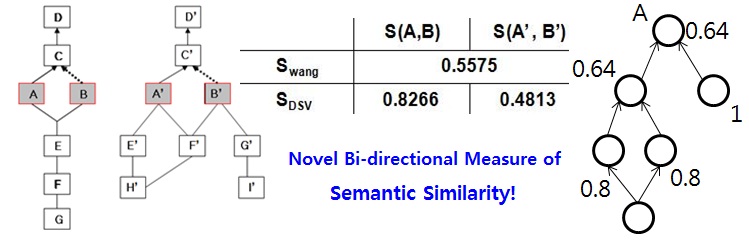
Current methods for measuring GO semantic similarities are limited to considering only the ancestor terms while neglecting the descendants. One can find many GO term pairs whose ancestors are identical but whose descendants are very different and vice versa. Moreover, the lower parts of GO trees are full of terms with more specific semantics. A novel method of bi-directional semantic similarities using the entire GO tree structure, including both the upper and lower parts outperformed other graph-based and information content-based methods.
Predicting microbial infection through gene expression host responses
 After this there is no truning back.
After this there is no truning back.
dCoxS, biological interpretation of paired gene sets
Previous differential co-expression analysis may not detect closely collaborating biological modules that are defined under biological knowledge, which are differentially co-expressed between conditions. We propose dCoxS algorithm that identifies conditional change of co-expression between gene sets. dCoxS detects differential co-expression of two gene sets in two steps. At first, dCoxS measures Interaction Score (IS), which represents expression similarity between the gene sets using sample pair-wise Renyi’s relative entropy, for each condition. Statistical test for the conditional differences of IS determines differential co-expression of gene sets across different conditions. We demonstrated the utility of dCoxS using simulation data and two publicly available microarray datasets. The analysis results indicated that set-wise differential co-expression analysis can be a powerful method for the understanding biological processes induced by the conditional changes.
BioLattice, biological interpretation of gene expression data
BioLattice is a mathematical framework based on concept lattice analysis for the biological interpretation of gene expression data. By considering gene expression clusters as objects and associated annotations as attributes and by using set inclusion relationships BioLattice orders them to create a lattice of concepts, providing an 'executive' summary of the experimental context. External knowledge resources such as Gene Ontology trees and pathway graphs can be added incrementally.
ArrayXPath - I & II, biological pathway-based analysis of gene expression data
ArrayXPath is a web-based service for mapping and visualizing microarray gene-expression data for integrated biological pathway resources using Scalable Vector Graphics. By integrating major bio-databases and searching pathway resources, ArrayXPath automatically maps different types of identifiers from microarray probes and pathway elements.
ChromoViz
Welcome to the visual world...
ChromoViz is an R package for multimodal visualization of microarray data,
DNA copy number alterations, cross-platform and cross-species comparisons,
and genomic non-expression data obtained from public databases onto chromosomes. (pdf)
DIB-C

DIB-C is a novel algorithm for identifying a subset of genes sharing a significant temporal expression pattern when replicates are used. Our algorithm requires no prior knowledge, instead relying on an observed statistic which is based on the first and second order differences between adjacent time-points. Here, a pattern is predefined as the sequence of symbols indicating direction and the rate of change between time-points, and each gene is assigned to a cluster whose members share a similar pattern. (pdf)
GOChase-II
Every inconsistency that has a begining has an end!
GOChase-II: correcting semantic inconsistencies from Gene Ontology-based annotations.
GOChase-I
GOChase is a set of web-based utilities to detect and correct the errors in GO-based annotations.
(1) GOChase-History resolves the whole modification history of GO IDs.
(2) GOChase-Correct highlights a ‘merged term’ and redirects it to the correct ‘target term’ into which the ‘merged term’ was merged. For a discarded (or ‘obsolete’) GO term, the nearest non-discarded parent term is recommended by GOChase. This function may be used by GO browsers such as AmiGo and QuickGO to fix the broken hyperlinks.
(3) A whole database (such as LocusLink) as a flat file can be input to GOChase, reporting the annotation errors and GOChase corrections.
(4) When one inputs a GO ID, GOChase will resolve all gene products annotated with the GO ID across all the major databases.Whatever you may imagine, you would see more...
KPGRN Genotyping Chip Experiment Analysis Viewer
KPGRN Genotyping Chip Experiment Analysis Viewer provides the web interface searching genotype/allele frequency, Pairwise LD/R^2 Statistic with window size 1M from 200 normal samples genotyped by Genome-Wide Human SNP Array 5.0 and can be downloaded bulk data( genotype/allele frequency, LD/R^2 Statsitic and CEL files)
GRIP
Genome research informatics pipeline.
TMA-TAB & TMA-RDF

TMA-TAB is a spreadsheet-based data format for TMA data submission to the TMA-OM supportive TMA database system. TMA-TAB was developed by simplifying, modifying and reorganizing classes, attributes and templates of TMA-OM into five entities: experiment, block, slide, core_in_block, and core_in_slide. Five tab-delimited formats (investigation design format, block description format, slide description format, core clinicohistopathological data format, and core result data format) were made, each representing the entities of experiment, block, slide, core_in_block, and core_in_slide. We implemented TMA-TAB import and export modules on Xperanto-TMA, a TMA-OM supportive database application, to facilitate data submission. Development and implementation of TMA-TAB and TMA-OM provide a strong infrastructure for powerful and user-friendly TMA data management.
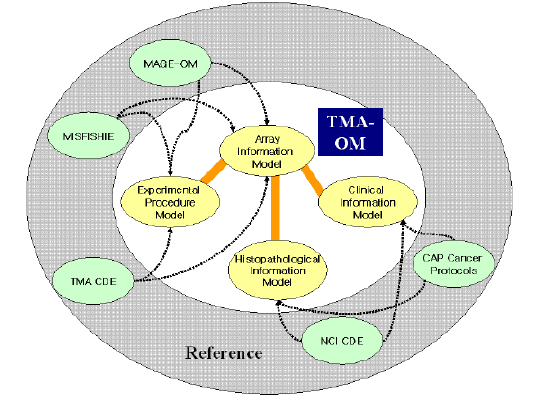
TMA-OM & Xperanto/TMA
The Tissue Microarray Object Model (TMA-OM) provides a comprehensive data model for storage, analysis, and exchange of clinical and histopathological information as well as TMA experimental data. We implemented a Web-based application, Xperanto-TMA, for TMA-OM, supporting data export in XML format conforming to the TMA data exchange specifications or the document type definition derived from TMA-OM. (pdf)
CaGe-OM
We developed a data model, CaGe-OM, to store and integrate data generated from microarray, proteomics and tissue microarray experiments performed on the same biological samples. The CaGe-OM can represent clinical and histopathological information as multiple functional genomics data for any type of cancer. (pdf)
FSGP: factor-specific generative patterns
Discovering significant and interpretable patterns from DNA microarray data.
Assuming underlying generative pattern. Defining factor-specific generative pattern and pattern distance. Genetic Algorithm to find the nearest factor-specific generative pattern of an observation. Determining significance:
MITree-K
MITree is a clusterin algorithm based on a geometric principle. Initially it was designed to be a binary hierarchical clustering algorithm for gene expression analysis. It is well described in Kim JH, Ohno-Machado L, Kohane IS. Unsupervised learning from complex data: the matrix incision tree algorithm. Pac Symp Biocomput 2001;:30-41. We also applied Evolution Strategy (i.e., a genetic algorithm) to the same problem. pdf Implementation of MITree is available here with a typical input data format example (Fisher's iris data set), where data starts from the 4th column and 2nd row and tab-dilimited ascii text file. Visualization and Evaluation of Clustering Structures for Gene Expression Data Analysis. Kim JH, Ohno-Machado L, Kohane IS. (2002) J Biomed Inform 2002;35(1):25-36
MITreeView
BioGPS: coming soon